Estrogen Data
A 2x2 factorial experiment
Gordon Smyth
16 August 2005
1. Aims
This case study illustrates more advanced linear modeling with Affymetrix single-channel microarrays. The popular 2x2 factorial design is considered. Use of Bioconductor annotation for Affymetrix arrays is illustrated. The case study goes on to consider significance tests using gene sets.
2. Required data
The estrogen data set is required for this lab and can be obtained from estrogen.zip. You should create a clean directory, unpack this file into that directory, then set that directory as your working directory for your R session using setwd() or otherwise. By typing dir() you should see eight .CEL files and three text files. On my computer, I see:
> setwd("C:/Gordon/www/bioinf/marray/bioc2005/_src/estrogen")
> dir()
[1] "estrogen.txt" "high10-1.cel" "high10-2.cel"
[4] "high48-1.cel" "high48-2.cel" "knownERgenes.txt"
[7] "low10-1.cel" "low10-2.cel" "low48-1.cel"
[10] "low48-2.cel" "predictedERgenes.txt"
To repeat this case study in full you will need to have the R packages affy, hgu95av2cdf, hgu95av2 and xtable installed.
3. Estrogen experiment
The data gives results from a 2x2 factorial experiment on MCF7 breast cancer cells using Affymetrix HGU95av2 arrays. The factors in this experiment were estrogen (present or absent) and length of exposure (10 or 48 hours). The aim of the study is the identify genes which respond to estrogen and to classify these into early and late responders. Genes which respond early are putative direct-target genes while those which respond late are probably downstream targets in the molecular pathway.
This data is from the estrogen data package on the Bioconductor website http://bioconductor.org/data/experimental.html. Rather than loading the data package here we simply using the nine basic data files from that package, in order to save storage space and to more closely mimic a real data analysis situation. The data set is discussed further by Scholtens [1,2] and in the Limma User's Guide.
4. Read the data
The first step in most analyses is to read the targets file which describes what RNA target has been hybridized to each array and, equally importantly, gives the names of the corresponding data files. The targets file is usually tab-delimited, but here it is white-space delimited.
library(limma)
targets <- readTargets("estrogen.txt", sep="")
targets
You should see
> targets
filename estrogen time.h
1 low10-1.cel absent 10
2 low10-2.cel absent 10
3 high10-1.cel present 10
4 high10-2.cel present 10
5 low48-1.cel absent 48
6 low48-2.cel absent 48
7 high48-1.cel present 48
8 high48-2.cel present 48
Now read the CEL file data into an AffyBatch object and normalize using RMA:
library(affy) library(hgu95av2cdf) abatch <- ReadAffy(filenames=targets$filename) eset <- rma(abatch)
Here eset is a data object of class exprSet.
It is usual and appropriate to check data quality before continuing your analysis. Due to brevity we will skip over this in this lab. A full set of quality assessment plots can be found at http://www.stat.berkeley.edu/~bolstad/PLMImageGallery/ under the title "estrogen". These plots show no significant quality problems with any arrays in this dataset.
5. Create a design matrix
We have four pairs of replicate arrays so we should estimate four parameters in the linear model. There are many valid ways to choose a design matrix, but perhaps the simplest is to make each column correspond to a particular treatment combination:.
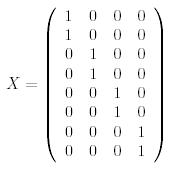
The four columns of the matrix correspond to
absent10, present10,
absent48 and present48,
respectively. Another way to specify the design matrix
is described in the Limma User's Guide.
This design matrix given above can be computed in R as follows:
f <- paste(targets$estrogen,targets$time.h,sep="") f <- factor(f) f design <- model.matrix(~0+f) colnames(design) <- levels(f) design
6. Fit the linear model
Now that we have defined our design matrix, fitting a linear model is as simple as:
fit <- lmFit(eset, design)
fit is an object of class
MArrayLM.
names(fit)
The fitted coefficents fit$coef from the model fit
are just the mean log-expression for each treatment
combination for each probe set. For this reason, this
choice of design matrix is called the group means
parametrization in the Limma User's
Guide.
7. Define a contrast matrix
The idea now is to use contrasts to make any comparisons of interest between the four treatment combination. Contrasts are linear combinations of parameters from the linear model fit.
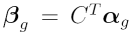
where 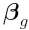 is a vector of contrasts
for gene
is a vector of contrasts
for gene 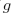 ,
, 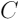 is the contrasts matrix, and
is the contrasts matrix, and
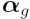 is a vector of coefficients
(estimated log fold changes), obtained from a linear
model fit.
is a vector of coefficients
(estimated log fold changes), obtained from a linear
model fit.
We will estimate three contrasts (so our contrasts matrix will have three columns). The first contrast is an estrogen effect at time 10 hours, the second as an estrogen effect at time 48 hours and the third is the time effect in the absence of estrogen. These are not all the comparisons which might have been made.

cont.matrix <- makeContrasts(E10="present10-absent10",E48="present48-absent48",Time="absent48-absent10",levels=design) cont.matrix
8. Extract the linear model fit for the contrasts
fit2 <- contrasts.fit(fit, cont.matrix) fit2 <- eBayes(fit2)
9. Assessing differential expression
We now use the function topTable to
obtain a list genes differentially expressed between
Estrogen-Present and Estrogen-Absent at time 10 hours,
followed by a list of genes differentially expressed
between Estrogen-Present and Estrogen-Absent at time 48
hours.
colnames(fit2) topTable(fit2,coef=1) topTable(fit2,coef=2,adjust="fdr") topTable(fit2,coef=2) topTable(fit2,coef=2,adjust="fdr")
The function decideTests() provides a variety of ways to assign statistical significance to the contrasts while controlling for multiple testing.
results <- decideTests(fit2) summary(results) vennDiagram(results)
10. Linking the gene lists to annotation information on the Internet
This section was prepared by James Wettenhall.
If the genes int the topTable have standard IDs
(e.g. UniGene or GenBank), then they can be linked with
external annotation information on the Internet. Load
the annotation package hgu95av2, which can
be obtained from http://bioconductor.org/data/metaData.html.
library(hgu95av2cdf) library(hgu95av2)
Now we obtain:
- the gene (probe-set) IDs (from the
AffyBatchobject,ab), - the gene symbols (from the
hgu95av2SYMBOLenvironment in thehgu95av2annotation package), - the gene names (from the
hgu95av2GENENAMEhgu95av2annotation package), and - the UniGene IDs (from the
hgu95av2UNIGENEhgu95av2annotation package).
geneIDs <- ls(hgu95av2cdf)
It is possible to extract the annotation information
from the appropriate R environments within the
annotation package (hgu95av2), and store
it in simple R data structures as follows:
# geneSymbols <- unlist(as.list(hgu95av2SYMBOL)) # geneNames <- unlist(as.list(hgu95av2GENENAME)) # unigene <- unlist(as.list(hgu95av2UNIGENE))
However we must be very careful, because in recent versions of Bioconductor annotation packages, some gene IDs can map to multiple gene names, multiple symbols and/or multiple unigene clusters. The method above, while appearing simple, also does not allow for the possibility that the gene names are stored in a different order in the annotation package from the gene IDs. We will therefore use some rather complicated-looking R code to extract the gene symbols, names and unigene IDs from the hgu95av2 environment. Multiple entries will be collapsed and separated by semicolons, e.g. if geneID 001 corresponds to gene names "Name1" and "Name2", these will be collapsed into "Name1; Name2".
geneSymbols <- as.character(unlist(lapply(mget(geneIDs,env=hgu95av2SYMBOL),
function (symbol) { return(paste(symbol,collapse="; ")) } )))
geneNames <- as.character(unlist(lapply(mget(geneIDs,env=hgu95av2GENENAME),
function (name) { return(paste(name,collapse="; ")) } )))
unigene <- as.character(unlist(lapply(mget(geneIDs,env=hgu95av2UNIGENE),
function (unigeneID) { return(paste(unigeneID,collapse="; ")) } )))
The key functions to note in the code above are
mget which extracts multiple annotation
strings from the appropriate annotation environmentand
lapply which applies our
multiple-entry-collapsing function to every element in
a list of annotation strings.
Now we abbreviate the gene names to a maximum length of 40 characters, for neat formatting in a table, and we extract the unigene ID from the string which contains "Hs" (for Homo sapiens) as well as the unigene ID.
geneNames <- substring(geneNames,1,40)
unigene <- gsub("Hs\\.","",unigene)
genelist <- data.frame(GeneID=geneIDs,GeneSymbol=geneSymbols,GeneName=geneNames,
UniGeneHsID=paste("<a href=http://www.ncbi.nlm.nih.gov/UniGene/clust.cgi?ORG=Hs&CID=",
unigene,">",unigene,"</a>",sep=""))
Now we recreate the toptable for the two contrasts considered earlier, E10="present10-absent10" and E48="present48-absent48" this time providing a hyperlink to the UniGene website for each gene in the toptable.
unigeneTopTableEst10 <- topTable(fit2,coef=1,n=20,genelist=genelist) unigeneTopTableEst48 <- topTable(fit2,coef=2,n=20,genelist=genelist)
library(xtable)
xtableUnigeneEst10 <- xtable(unigeneTopTableEst10,display=c("d","s","s","s","s","g","g","g","e","g"))
xtableUnigeneEst48 <- xtable(unigeneTopTableEst48,display=c("d","s","s","s","s","g","g","g","e","g"))
cat(file="estrogenUniGeneE10.html","<html>\n<body>")
print.xtable(xtableUnigeneEst10,type="html",file="estrogenUniGeneE10.html",append=TRUE)
cat(file="estrogenUniGeneE10.html","</body>\n</html>",append=TRUE)
cat(file="estrogenUniGeneE48.html","<html>\n<body>")
print.xtable(xtableUnigeneEst48,type="html",file="estrogenUniGeneE48.html",append=TRUE)
cat(file="estrogenUniGeneE48.html","</body>\n</html>",append=TRUE)
The display argument to the xtable
function is used to specify the format of text or
numbers displayed in cells of the HTML table:
| Format code | Meaning |
|---|---|
| d | Decimal (base ten) integer, e.g. 48 |
| s | Character string, e.g. "Block" |
| g | General real floating-point number, e.g. 8.25 |
| e | Floating point number in exponent format, e.g. 1.02E-05 |
11. Gene Set Enrichment
This section was prepared by James Wettenhall.
11.1 Introduction to Gene Set Analysis
In this lab, we move beyond the analysis of individual genes, and consider sets of genes in microarray experiments. Another approach is to form gene sets based on a priori knowledge of common biological features shared by the genes. We consider a particular approach called gene set enrichment. We begin with a known set of genes and then test whether this set as a whole is differentially expressed in a microarray experiment. This type of test is useful when comparing one's microarray data with that of previous authors who have performed similar microarray experiments, because the lists of most differentially expressed genes reported by the previous authors can be regarded as a "gene set" and tested to determine whether the genes are also differentially expressed in the current context.
Gene set testing was introduced by Mootha et al [5] and Lamb et al [6] in 2003. Mootha et al define the concept of a gene set enrichment test. For a given set of genes, one can test whether the set as a whole is up-regulated, down-regulated or differentially expressed with individual genes possibly going in either direction. Sometimes performing the traditional differential expression analysis of individual genes will yield no statistically significant results, but there may be stronger evidence for differential expression of gene sets.
Now we turn our attention to tests for differential expression involving a set of genes. Mootha et al. [5] and Lamb et al. [6] made this methods popular in 2003. We will use a "gene set enrichment test", which is closely based on the one defined by Mootha et al. The gene set test can be used to test whether previous author's lists of differentially expressed genes are also differentially expressed in a current experiment similar to that of the previous authors. Another possible application is to try to find differential expression in microarray experiments which show no strong differential expression when testing for individual differentially expressed genes, but they might show more evidence of differential expression when testing a predefined set of genes. Defining a useful gene set for this sort of analysis is not always trivial. One possibility is to use a set of genes which share common gene ontologies, i.e. choose a set of genes which are all associated with GOs below a certain node in the GO DAG (Directed Acyclic Graph). We will begin with some artificial examples to illustrate the concept of gene set tests with a small number of made-up t-statistics. Then we will use two sets of genes thought to be regulated by the Estrogen Receptor (ERalpha) to demonstrate testing for differential expression of gene sets in the Estrogen data set.
The geneSetTest function in the limma
package [8] is described in its help file, reproduced
below:
We now demonstrate the use of the
geneSetTest function in limma
using a miniscule set of artifical made-up
t-statistics, where as usual, a positive t statistic
means that a gene is up-regulated (i.e. expressed more
highly in a condition of interest), whilst a negative
value means that the gene is down-regulated. A
t-statistic close to zero means that the gene is not
differentially expressed.
In the first example we use, the artificial
t-statistics will range from -9 to 9, and we will
select a set of three genes for the test, those with
t-statistics of -8, -6 and -5, i.e. we will use the
2nd, 4th and 5th t-statistics from our vector of
artificial t-statistics. If these t-statistics
represented real genes, all three genes would show
strong evidence of differential expression
(down-regulation) individually, so they should
certainly show strong evidence of differential
expression as a set as well. The value returned by
limma's geneSetTest function is a
p-value.
library(limma) sel <- c(2,4,5) stat <- -9:9 stat[sel] geneSetTest(sel,stat,nsim=100) geneSetTest(sel,stat,ranks.only=TRUE)
If we did a test for up-regulation of the set, we would expect a large p-value (low evidence of up-regulation):
geneSetTest(sel,stat,alternative="greater",nsim=100)
On the otherhand, a test for down-regulation should give a small p-value:
geneSetTest(sel,stat,alternative="less",nsim=100)
11.2 Gene Set Analysis Example
We will again use the Estrogen data set through the
fit2 linear model fit object.
We will use two sets of genes which are thought to be ER-regulated, i.e. regulated by the Estrogen Receptor alpha. The first set (Jin et al [4]) contains genes which have been experimentally verified to be ER-regulated and the second set (O'lone et al [7]) contains a large list of genes which are predicted to be ER-regulated by a model (so they may or may not be ER-regulated).
These gene sets (particularly the first one) should be differentially expressed between the breast cancer cells with estrogen reintroduced and the serum-starved breast cancer cells with no estrogen, because in the cells reintroduced to estrogen, the estrogen receptors (ERs) will bind the estrogen and as a result become activated, gaining the ability to regulate gene expression in the cells, hence resulting in differential expression between the cells with and without estrogen.
The data required for this exercise is available from knownERgenes.txt and predictedERgenes.txt. Read the gene lists into R:
known <- read.delim("knownERgenes.txt",as.is=TRUE)
knownERgenes <- known$UGCluster
predicted <- read.delim("predictedERgenes.txt",as.is=TRUE)
predictedERgenes <- predicted$UGCluster
library(hgu95av2)
unigene <- unlist(as.list(hgu95av2UNIGENE))
knownERgenesOnChip <- match(knownERgenes,unigene)
knownERgenesOnChip <- knownERgenesOnChip[!is.na(knownERgenesOnChip)]
predictedERgenesOnChip <- match(predictedERgenes,unigene)
predictedERgenesOnChip <- predictedERgenesOnChip[!is.na(predictedERgenesOnChip)]
Now we will use the moderated t-statistics calculated previously for the comparison of estrogen present and estrogen absent 10 hours after the estrogen was reintroduced into the cells. We try all three types of tests - "two-sided", "greater" (for genes up-regulated in the sample with estrogen present), and "less" (for genes down-regulated in the sample with estrogen present). We expect to get better (smaller) p-values for the known ER-regulated genes than for the predicted ER-regulated genes. The recent review of Estrogen-repressed genes in breast cancer cells by Zubairy and Oesterreich [9] suggests that the majority of genes regulated by estrogen receptors are actually repressed (down-regulated), so we should expect a lower p-value for the "less" test (at least for the known ER-regulated genes).
geneSetTest(knownERgenesOnChip,fit2$t[,1],"two.sided") geneSetTest(knownERgenesOnChip,fit2$t[,1],"greater") geneSetTest(knownERgenesOnChip,fit2$t[,1],"less") set.seed(0) geneSet <- sample(predictedERgenesOnChip,length(knownERgenesOnChip)) geneSetTest(geneSet,fit2$t[,1],"two.sided") geneSetTest(geneSet,fit2$t[,1],"greater") geneSetTest(geneSet,fit2$t[,1],"less") geneSet <- sample(predictedERgenesOnChip,length(knownERgenesOnChip)) geneSetTest(geneSet,fit2$t[,1],"two.sided") geneSetTest(geneSet,fit2$t[,1],"greater") geneSetTest(geneSet,fit2$t[,1],"less") geneSet <- sample(predictedERgenesOnChip,length(knownERgenesOnChip)) geneSetTest(geneSet,fit2$t[,1],"two.sided") geneSetTest(geneSet,fit2$t[,1],"greater") geneSetTest(geneSet,fit2$t[,1],"less")
Acknowledgements
Thanks to Denise Scholtens and Robert Gentleman for providing the estrogen data http://bioconductor.org/data/experimental.html. Thanks to Ben Bolstad for a quality analysis of this data.
Thanks to Hui Tang and Terry Speed for finding the known and predicted ER-regulated gene sets (Jin et al. [4] and O'lone et al. [7]). Thanks to James Wettenhall for material on linking to internet annotation and for the gene set worked example.
References on factorial studies
- Scholtens D, Gentleman R. Estrogen 2x2 Factorial Design. http://bioconductor.org/repository/devel/vignette/factDesign.pdf
- Scholtens D, Miron A, Merchant FM, Miller A, Miron PL, Iglehart JD, Gentleman R. Analyzing Factorial Designed Microarray Experiments. Journal of Multivariate Analysis. To appear.
- Smyth, G. K. (2005). Limma: linear models for microarray data. In: Bioinformatics and Computational Biology Solutions using R and Bioconductor, R. Gentleman, V. Carey, S. Dudoit, R. Irizarry, W. Huber (eds.), Springer, New York.
- Smyth, G. K., Thorne, N. P. and Wettenhall J. (2005) limma: Linear Models for Microarray Data User's Guide. http://bioinf.wehi.edu.au/limma (See the estrogen case study.)
References on gene sets
- Jin VX, Leu YW, Liyanarachchi S, Sun H, Fan1 M, Nephew1 KP, Huang T.H. and Davuluri DV (2004). Identifying estrogen receptor {alpha} target genes using integrated computational genomics and chromatin immunoprecipitation microarray. Nucleic Acids Research 32(22): 6627-6635
- Lamb J, Ramaswamy S, Ford HL, Contreras B, Martinez RV, Kittrell FS, Zahnow CA, Patterson N, Golub TR, Ewen ME (2003). A mechanism of cyclin D1 action encoded in the patterns of gene expression in human cancer. Cell; 114(3):323-34.
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle, M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman BM, Lander ES, Hirschhorn JN, Altshuler D, and Groop LC (2003). PGC-1alpha Responsive Genes Involved in Oxidative Phosphorylation are Coordinately Downregulated in Human Diabetes, Nature Genetics 34(3):267-73.
- O'lone R, Frith MC, Karlsson EK, Hansen U (2004). Genomic Targets of Nuclear Estrogen Receptors. Molecular Endocrinology 18(8): 1859-1875
- Zubairy S, Oesterreich S (2005). Estrogen-repressed genes -- key mediators of estrogen action? Breast Cancer Res. 2005;7(4):163-4.